在日常生活中,我們難免遇到細長毛線、繩索或耳機線糾纏打結的困擾。理論計算的結果顯示,任何足夠長度的鏈狀物只要有充分的時間被甩動及堆疊交錯,幾乎都會不可避免地陷入打結狀態。在生物系統中,最常見的紐結構型是環狀雙股螺旋DNA,通過拓樸異構酶(topoisomerase)的作用,DNA紐結可以選擇性地被解開而產生不同的拓樸構型。如同DNA,蛋白質亦是線性聚合物,其是透過胜肽鍵連接20 種不同胺基酸排列組合而成。不過,從蛋白質多肽鏈骨架所延伸出來的側鏈基團提供蛋白質序列在一維空間中高度物理化學性質變異性,因而與DNA及其他容易形成紐結的長鏈高分子有本質上的差異。也因為如此,除了日漸受到矚目的天然無序蛋白質(intrinsically disordered proteins, IDPs),大多數的蛋白質序列會透過不同的螺旋與平板二級結構在空間中形成特定的排列組合,進而堆疊成緊緻的三維結構以執行特定的生物功能。蛋白質分子如何能高效率地在極短時間內,從近乎天文數字種可能性的排列組合中,尋找到最穩定,即最低自由能的三維構型,正是所謂的「蛋白質摺疊問題」。該問題隨著實驗技術的精進以及理論計算能力的躍進,在近年來有重大的突破:對於較小的單區域(single domain)蛋白質,實驗與理論計算可以觀察共同時間尺度中的結構資訊而互相驗證。當特定蛋白質因為受到外在壓力、家族性遺傳突變改變胺基酸序列,或是因為不當的轉譯後修飾(post-translational modifications, PTMs)而無法維持原有的天然構型,甚至堆疊形成類澱粉纖維化組織(amyloid fibril),產生錯誤摺疊的構型會導致生理功能的喪失,甚至造成疾病。與蛋白質錯誤摺疊最直接相關的常見疾病包括阿茲海默症、帕金森氏症、亨丁頓氏症、漸凍人以及狂牛症等神經衰退疾病。
由於蛋白質普遍具有構型緊緻摺疊的特性,即便Richardson早在1977年利用拓樸分類法分析已知蛋白晶體結構便發現蛋白碳酸酐酶(carbonic anhydrase, CA)具有形成三葉型結(trefoil knot)的31結特殊拓樸構型[1],但長久以來,生化學家仍舊認為蛋白質應有別於一般線性高分子而不會打結。值得一提的是,就數學紐結理論的定義而言,紐結僅存在於封閉無開口的路徑,而蛋白質在生合成後是以線性多肽鏈的形式存在,因此蛋白紐結是具有開放末端的。嚴謹而言,蛋白質紐結若要符合數學上的紐結定義,需將氮(N)與碳(C)兩端進行虛擬連接來達成(見圖一A)。由於線性多肽鏈開放端的存在,蛋白質紐結的檢測需要依賴空間中開放曲線投影簡化的數學分析方法。Mansfield於1994年提出了一套數學分析方法來系統性地分析當時蛋白質結構資料庫(protein data bank, PDB)僅有的400多個蛋白質結構後同樣發現Richardson當年當年指出CA結構中的31結,而這是當時唯一被發現的打結蛋白結構[2]。
隨著二十世紀末人類基因體序列的建立,各國研究機構開始提出不同物種的結構基因體計劃(structural genomics projects)試圖透過大規模地鑑定蛋白質結構,進而推導基因序列與蛋白質三度空間結構的關係,同時希望透過系統性地分類所有物種的蛋白質結構及拓樸類型,來建立生物演化過程中所產生的蛋白質構型原件。在結構生物學家近二十年的共同努力下,PDB目前已儲存逾十四萬筆生物巨分子的原子模型。同時,理論學家也開發了不同的蛋白質紐結偵測語法,系統性地搜尋不同類型的蛋白質紐結(見圖一A),其中波蘭華沙大學Sulkowska實驗室所開發的KnotProt(http:/knotprot.cent.uw.edu.pl/)會週期性地更新及統計PDB中具有紐結構型的蛋白質結構[3]。根據KnotProt,截至2018年七月底,PDB已記錄有1053個具有紐結構型的蛋白質結構,包括三葉型(31)結、八字型(41)結、戈迪安(Gordian, 52)結和最複雜的碼頭工人(Stevedore, 61)結。其中具有31結的CA家族佔最大比例,其次是參與RNA甲基轉移催化反應且同樣含有31結的SPOUT家族。另一個較具代表性也比31結更為複雜的打結蛋白家族則為帶有52結的泛素碳端水解酶(ubiquitin C-terminal hydrolases, UCHs)[4]。
蛋白紐結構型是否是為了因應功能上的需求而在演化中產生的?
透過一系列的結構比對分析可以發現,蛋白紐結構型在不同家族的演化過程中,多半在一開始便被保存下來。以SPOUT和UCH家族為例,目前已知的所有家族成員的分子結構中都含有共同的31結和52結構型。雖然SPOUT家族成員已有相當多的分支,彼此之間的胺基酸序列相似度也有限,但是所有成員均透過相同的特殊31結構型辨識反應基質S-腺苷-L-高半胱氨酸(S-adenosyl-L-homocysteine, SAH;見圖一B)[5]。同樣的,僅在真核生物中存在的UCH家族成員,從酵母菌到人類都具有共同的52結構型,而其N端也是與泛素結合的重要結構辨識組件之一[6]。在目前已知的紐結蛋白家族中,脆弱擬桿菌的N-琥珀酰基-L-鳥氨酸轉氨甲酰酶(Bacteroides fragilis N-succinyl-l-ornithine transcarbamylase, BfSOTC)和野油菜黃單胞菌的N-乙酰基-L-鳥氨酸轉氨甲酰酶(Xanthomonas campestris N-acetyl-l-ornithine transcarbamylase, XcAOTC)是唯一由未打結的鳥氨酸轉氨甲酰酶超級家族中分支出來的紐結蛋白成員。如同其他紐結蛋白,XcAOTC和BfSOTC的31結結構也會參與辨識精氨酸(arginine)及尿素生合成前驅物N-乙酰基-L-鳥氨酸轉氨甲酰酶(ARO)。XcAOTC能有效地辨識ARO上的乙酰基進行高選擇性的酵素反應;相反的,大腸桿菌的L-鳥氨酸轉氨甲酰酶(EcOTC)則無法有效辨識ARO額外的乙酰基(見圖一B)。OTC超級家族也就成為了解蛋白紐結在演化上功能與結構相關性的難得的自然生物系統。
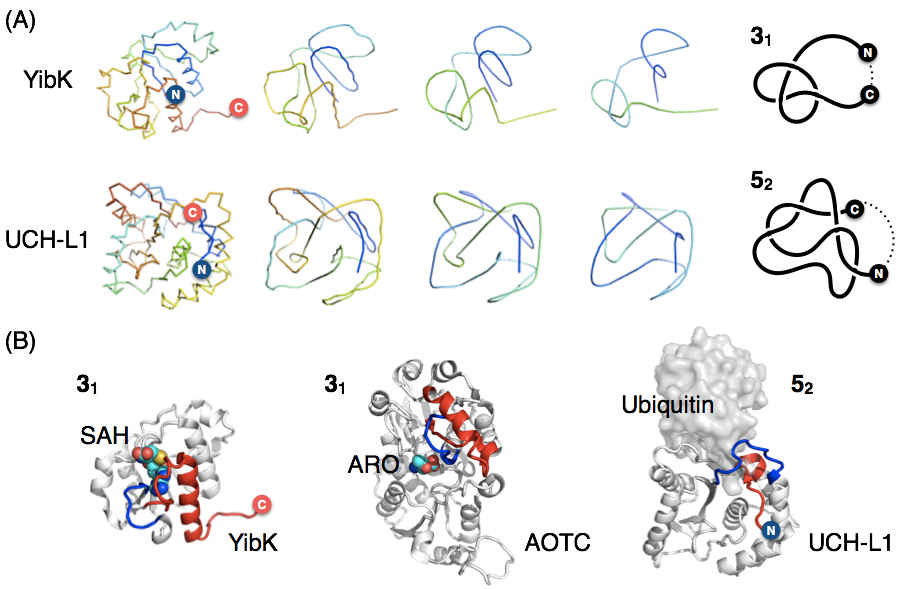
(圖一)蛋白質三維結構中紐結構型的偵測以及蛋白紐結參與基質辨識之範例。大多數蛋白質的三維結構錯綜複雜,不容易以肉眼判斷骨架摺疊之拓樸構型是否含有紐結。其中一個蛋白紐結偵測方式是透過固定蛋白質分子構型的N端與C端坐標後,將骨架路徑圓滑簡化,但交錯的路徑不能跨越彼此。前交通大學黃鎮剛教授引進該計算法所發表之 pKNOT網站[7]可以讓使用者上傳已知之蛋白質分子結構座標,協助簡化骨架路徑,進而透過視覺辨識紐結的存在。圖A以本實驗室所研究之具有31結的流感嗜血桿菌(Haemophilus influenzae)RNA甲基轉移酶YibK及52結的人類UCH-L1作為範例,顯示骨架路徑圓滑程序之計算結果,同時在最右方圖示將N端與C端以虛線連結後形成封閉路徑以符合數學紐結理論定義之圖像描述。許多蛋白紐結結構會直接參與辨識基質。圖B將三個具代表性的紐結蛋白:YibK、AOTC 及UCH-L1以卡通模式呈現並將參與紐結構型的環狀構型顯示為藍色並將穿過該環狀構型的骨架路徑顯示為紅色,並將其SAH及ARO分子構型以淺藍、深藍、紅色及黃色球形顯示碳、氮、氧及硫原子;而UCH-L1所辨識之泛素分子則以半透明表面模型顯示。
透過多參數物理化學分析方法建立紐結蛋白摺疊機制理論
自從紐結蛋白在結構資料庫中陸續被發現後,實驗與理論學家開始探討紐結蛋白的打結機制。透過不同空間與時間尺度的理論計算,有三種蛋白質打結機制被提出,其中包括直接穿線(direct threading)、滑動打結(slip-knotting)和捕鼠器翻蓋(mouse trapping)等機制。針對紐結蛋白摺疊的實驗資訊主要源起於英國劍橋大學Sophie Jackson實驗室於2005年開始對於具有31結的流感嗜血桿菌(Haemophilus influenzae)RNA甲基轉移酶YibK與大腸桿菌甲基轉移酶YbeA的一系列研究工作。該團隊發現這兩個SPOUT家族成員雖然有相近的31結構型,但是其摺疊路徑相去甚遠[8]。有趣的是,Jackson等人指出,YibK與YbeA在外加大量尿素導致蛋白質變性、失去二級與三級結構時,仍舊處於打結的狀態[9]。此外,YibK與YbeA可以在完成生合成離開核糖體之後,自發性地完成打結狀態;外加伴隨蛋白酶(molecular chaperone)GroEL/GroES則會大幅加速這兩個打結蛋白的摺疊速率[10]。換言之,有別於DNA需要借由拓樸異構酶的協助以達成拓樸紐結構型,紐結蛋白可以自發性地完成特殊且緊緻的紐結狀態。
自2010起,本實驗室在人類前鋒科學研究計劃(Human Frontier Science Program)及科技部的資助下,開始系統性地研究紐結蛋白的摺疊機制,透過多參數生物物理化學方法,比較分析不同類型紐結蛋白以及同一家族成員的紐結蛋白摺疊熱力學與動力學參數,進而建立各個紐結蛋白構型摺疊路徑自由能景觀(folding free energy landscape)。其中包括具有最小31結的嗜熱菌(Methanocaldococcus jannaschii )蛋白MJ0366[11]及具有最複雜61結的惡臭假單胞菌鹵酸脫鹵素酶(Pseudomonas putida haloacid dehalogenase, DehI)[12]。加上針對人類UCH家族成員UCH-L1、UCH-L3、UCH-L5及BAP1的一系列比較性分析,我們發現雖然不同類型的紐結蛋白質對於外加物理化學刺激(例如溫度以及化學物質)的耐受度各有不同,但所有紐結蛋白在展開構型的過程中,都會出現明顯的中間狀態(intermediate state, I)。有些紐結蛋白可以透過單一路徑形成一個簡單的中間狀態,有些紐結蛋白(例如DehI)會在同一路徑上出現兩種不同的中間狀態[12],其他紐結蛋白(例如UCH-L1)則會透過兩條平行路徑形成不同的中間狀態,並各自連結自然狀態(native state, N)與變性狀態(denatured state, D)[13]。蛋白質摺疊過程中出現顯著中間狀態的結果顯示,紐結蛋白構型摺疊路徑自由能景觀充滿能量陷阱,導致紐結蛋白從高能量的變性狀態轉變為低能量的自然狀態過程中充滿挫折(frustration)。這個觀察與理論計算理論計算提出紐結的形成是紐結蛋白摺疊過程的速率限制步驟(rate-limiting step)的假說是一致的。
與帕金森氏症息息相關的UCH-L1
具有52結的UCH-L1是人類腦神經元中表現量最高的蛋白質之一,UCH-L1經常與α-突觸核蛋白(α-synuclein)一起出現在帕金森氏症患者腦部的路易氏體(Lewy body)。針對帕金森氏症家族性遺傳I93M對人類UCH-L1造成的結構、動態與功能的影響進行分子層面的分析。雖然UCH-L1野生種(wild type)與帕金森氏症家族遺傳I93M突變體的晶體結構並無明顯差距,本實驗室透過液態核磁共振光譜(nuclear magnetic resonance, NMR)顯示I93M突變造成對UCH-L1酵素反應中心以及周遭的化學環境產生顯著影響[14]。本實驗室與Sophie Jackson實驗室合作並結合NMR、蛋白質自體螢光以及圓二色旋光(CD)光譜分析發現UCH-L1在自然狀態時即存在許多部分展開構型(partially unfolded forms, PUFs),而其結構特性與透過外加尿素引發化學變性反應產生中間狀態有極高的相似度[13]。雖然蛋白質摺疊反應中間狀態的分子構型決定不易,但是透過螢光光譜分析可以確知UCH-L1在透過化學物質驅使構型展開的過程中會經由兩條不同路徑產生不同的中間狀態(I及I’),進而完全失去其二級及三級結構,成為變性狀態(見圖二)。值得一提的是,NMR、螢光及CD光譜只能反應不同的結構表徵,而且這些結構表徵多半為局部結構資訊,因此即使有這些不同的結構資訊,闡述蛋白質構型產生的摺疊路徑依舊有瞎子摸象的不完整性。本實驗室在近日透過國家同步輻射研究中心之小角度X光散射(small angle X-ray scattering, SAXS)分析方法以及多角度靜態光散射結合線上分子篩層析分離技術(size exclusion chromatography coupled multi-angle light scattering, SEC-MALS),證明 UCH-L1的中間狀態其實是具有高度結構亂度的二聚體與四聚體。在移除化學變性物質之後,中間狀態二聚體與四聚體可以回復到以單聚體形式存在的自然狀態[15]。但是當UCH-L1出現I93M突變之後,外加尿素會導致UCH-L1聚集形成非常大且無序的聚集沈澱;在高蛋白濃度的情況下,即便事後移除外加尿素也無法有效將UCH-L1帶回原先的自然狀態。該發現顯示UCH-L1其實存在於亞穩定狀態(metastable state),任何突變及轉譯後修飾都很容易破壞其穩定性而引發錯誤摺疊與聚集沈澱的現象。這個發現或許可以解釋為何UCH-L1經常與α-synuclein一起堆疊在路易氏體。
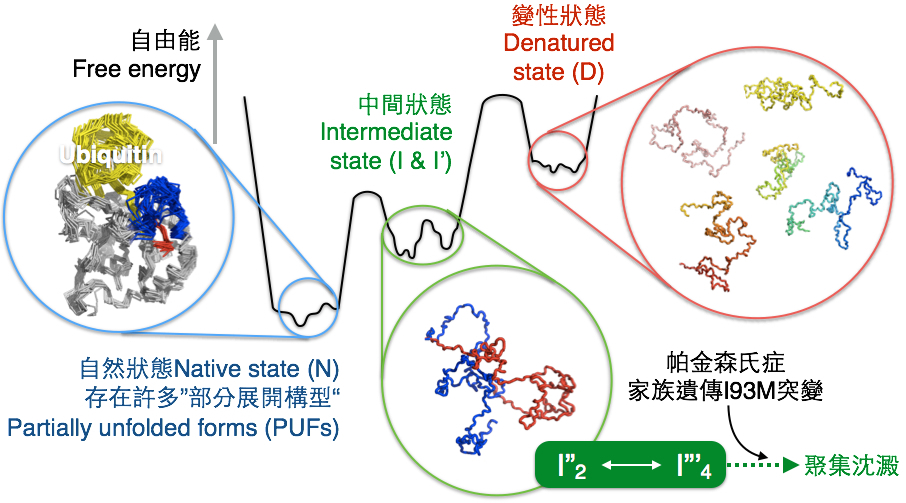
(圖二)透過多重物理化學參數熱力學與動力學分析方法建立人類UCH-L1構型摺疊路徑自由能景觀。
紐結提供蛋白質極高的拓樸力障與蛋白酶體拔河抵抗展開構型的外力
在比較人類UCH-L1、UCH-L3與UCH-L5的摺疊熱力學與動力學的過程中,我們發現UCH-L5具有極為緩慢的自發性構型展開速率(intrinsic unfolding rate)。一般而言,穩定的蛋白質具有較緩慢的自發性構型展開速率,而其構型在自然狀態下也相對有較少的動態與結構變異。但是透過比對UCH-L5的NMR氫氘交換與螢光實驗數據,我們發現UCH-L5在自然狀態下存在比UCH-L1及UCH-L3都還要更加明顯的區域性結構熱擾動(thermal fluctuation),但其整體熱力學穩定性卻比另外兩個UCH家族成員要高出許多,其構型展開動力學反應速率也比UCH-L1慢了近一萬倍。從熱力學的角度來看,系統的穩定性除了透過鍵結(焓)穩定構型之外,也可以透過增加系統亂度(熵)來降低自由能。由於UCH-L5的泛素C端水解功能需要透過與蛋白酶體(proteasome)上的泛素受器(Rpn13)結合之後才會被活化,此外,UCH-L5也需與蛋白酶體競爭同一個被泛素化的基質,UCH-L5基本上是與持續透過水解三磷酸腺苷(ATP)產生外力展開泛素化基質的蛋白酶體進行拔河。因此我們推測UCH-L5的52結以及其極為緩慢的自發性構型展開速率應該會提供UCH-L5較高的力學穩定性。
為了證明這個假說,我們利用大腸桿菌的蛋白酶體ClpXP作為模式系統,測試UCH家族成員的力學穩定性。透過蛋白質工程技術,將帶有11個胺基酸的ssrA序列(AANDENYALAA;圖中銜接於UCH碳端的紅色片段)標記在UCH的C端, ClpX環狀六聚體可以辨識ssrA序列,並透過水解ATP產生外力展開基質之立體構型成為線性多肽鏈,接著被送入由雙層環狀七聚體所形成的封閉空間進行單一方向且具高度持續性(processivity)的水解反應將基質分解;換言之,ClpP必須有效地透過水解ATP產生脈衝式的拉力,將具有C端ssrA標記的基質蛋白展開成線性多肽鏈以跨越ClpXP酵素動力學的速率限制步驟。ClpXP的酵素動力學以及ATP水解所產生之力學展開基質蛋白分子機制在文獻均有詳細記載,其中,具備高度熱穩定性(變溫溫度Tm > 85 °C)及化學穩定性之綠色螢光蛋白(GFP)是最常被用為標準基質的選擇之一。相較於GFP,人類UCH-L1、UCH-L3、UCH-L5及BAP1在45-55 °C以及相對少量的化學變性物質(例如尿素)便會逐漸失去其分子構型的穩定性。即便如此,我們發現UCH-L1與UCH-L5受到ClpXP外力展開結構進而被水解的反應速率較GFP慢了一百至一萬倍,顯示這兩個UCH分子具有極高的力學穩定性[16]。我們更進一步透過剪短UCH-L1的N端十一個胺基酸以解開其52紐結構型(UCH-L111;請參見圖ㄧB之UCH-L1分子模型,其氮端的數個胺基酸形成52紐結構型的元件之一)而顯著加速ClpXP的反應速率證明UCH-L1的拓樸力障是導致ClpX無法有效展開其三維空間分子構型的主因之一。UCH-L5因此成為現今文獻報導中最能有效抑制ClpXP施加外力展開基質構型的蛋白質。
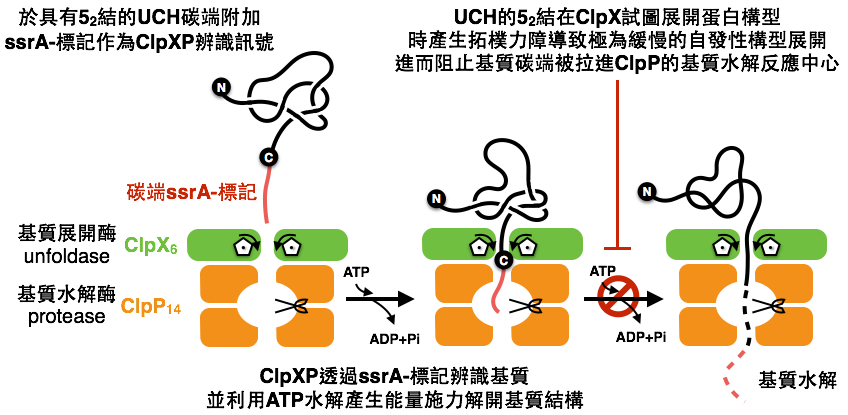
(圖三)利用大腸桿菌之ClpXP蛋白質展開酶/水解酶錯合物評量52紐結構型對於UCH家族蛋白質成員力學穩定性貢獻實驗設計示意圖。
結語
紐結蛋白質存在於自然界各個物種之中,其三維空間構型與拓樸各有不同。透過分析現今上千筆紐結蛋白的結構與相似家族成員的序列比對,應該可以窺見偏好紐結形成的序列特性,然而這項生物資訊分析工作仍有待專家嘗試。從蛋白質形成紐結的機制分析研究工作來看,透過本實驗室與其他理論與實驗學家的共同努力,我們對於紐結蛋白的形成的分子機制,生物意義以及對應不同物理化學變數(包含溫度、化學能以及外力)的穩定性已逐漸有所了解。然而,如同本文所述,即便透過各式各樣生物物理分析方法取得關於紐結蛋白質的結構資訊,闡述蛋白質構型產生的摺疊路徑依舊有瞎子摸象的不完整性。為了能有更全面的實驗資訊提供理論計算做交叉比對,本實驗室積極引入新穎實驗技術提升蛋白質結構分析的時間與空間解析度。中央研究院內的生物物理核心設施,高磁場核磁共振光譜核心設施以及生化所質譜核心設施最近引進的氫氘交換質譜分析方法,提供了最佳的技術支援。我們期望在未來能借重如此完善的研究設施對於紐結蛋白質的生物功能與拓樸紐結的相關性獲得更深入的認知,進而了解大自然為何大費周章演化出如此複雜的結構。
參考文獻
[1] J.S. Richardson, b-Sheet topology and the relatedness of proteins, Nature, 268 (1977) 495-500.
[2] M.L. Mansfield, Are there knots in proteins?, Nat. Struc. Biol., 1 (1994) 213-214.
[3] M. Jamroz, W. Niemyska, E.J. Rawdon, A. Stasiak, K.C. Millett, P. Sulkowski, J.I. Sulkowska, KnotProt: a database of proteins with knots and slipknots, Nucleic Acids Res., 43 (2015) D306-314.
[4] S.E. Jackson, A. Suma, C. Micheletti, How to fold intricately: using theory and experiments to unravel the properties of knotted proteins, Curr. Opin. Struct. Biol., 42 (2017) 6-14.
[5] K.L. Tkaczuk, S. Dunin-Horkawicz, E. Purta, J.M. Bujnicki, Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases, BMC Bioinformatics, 8 (2007) 73.
[6] S.-T.D. Hsu, Folding Dynamics and Structural Basis of the Enzyme Mechanism of Ubiquitin C-Terminal Hydroylases, in: A. Svendsen (Ed.) Understanding enzymes – function, design, engineering and analysis, Pan Stanford Publishing, Singapore, 2016, pp. 167-202.
[7] Y.L. Lai, C.C. Chen, J.K. Hwang, pKNOT v.2: the protein KNOT web server, Nucleic Acids Res., 40 (2012) W228-231.
[8] A.L. Mallam, S.E. Jackson, Probing nature’s knots: the folding pathway of a knotted homodimeric protein, J. Mol. Biol., 359 (2006) 1420-1436.
[9] A.L. Mallam, J.M. Rogers, S.E. Jackson, Experimental detection of knotted conformations in denatured proteins, Proc. Natl. Acad. Sci. U. S. A., 107 (2010) 8189-8194.
[10] A.L. Mallam, S.E. Jackson, Knot formation in newly translated proteins is spontaneous and accelerated by chaperonins, Nat. Chem. Biol., 8 (2012) 147-153.
[11] I. Wang, S.-Y. Chen, S.-T.D. Hsu, Unraveling the folding mechanism of the smallest knotted protein, MJ0366, J. Phys. Chem. B, 119 (2015) 4359-4370.
[12] I. Wang, S.-Y. Chen, S.-T.D. Hsu, Folding analysis of the most complex Stevedore’s protein knot, Sci. Rep., 6 (2016) 31514.
[13] S.-C. Lou, S. Wetzel, H. Zhang, E.W. Crone, Y.-T. Lee, S.E. Jackson, S.-T.D. Hsu, The knotted protein UCH-L1 exhibits partially unfolded forms under native conditions that share common structural features with its kinetic folding intermediates, J. Mol. Biol., 428 (2016) 2507-2520.
[14] S.M. Kumar, P.C. Lyu, S.-T.D. Hsu, Structural perturbation of the Parkinson’s disease-associated I93M mutation in human UCH-L1 revealed by solution state NMR spectroscopy, Chinese J. Magn. Reson., 32 (2015) 329-340.
[15] Y.C. Lee, S.D. Hsu, A natively monomeric deubiquitinase UCH-L1 forms highly dynamic but defined metastable oligomeric folding intermediates, J. Phys. Chem. Lett., 9 (2018) 2433-2437.
[16] M.K. Sriramoju, Y. Chen, Y.C. Lee, S.-T.D. Hsu, Topologically knotted deubiquitinases exhibit unprecedented mechanostability to withstand the proteolysis by an AAA+ protease, Sci. Rep., 8 (2018) 7076.