人類社會的運作,主要由能源所驅動。隨著經濟的發展,能源使用量日益增加,為了避免有限的能源儲藏被消耗殆盡,現今全球專注的焦點為開發能源轉換新技術。在各式再生能源技術中,依台灣的地理環境來考量,太陽能及風能發電為最具經濟效益及永續性的能源。(關於染料敏化太陽能電池的發展,請參考本院週報第483期「知識天地」,林彥多博士與周大新研究員之報導:「染料敏化太陽能電池的新發展」)。然而,此兩種再生能源並非基載電力來源,發電量受到環境影響很大。在這類發電機組大量架設前,要考量的其中一個重要因素為,如何在發電端及用電端中間設置能源儲存裝置,用於調節尖峰及離峰之發電及負載量,以避免電力網絡的崩潰。針對此關鍵點,一般常使用的是電池堆。以現今技術成熟度及符合經濟和空間最大化來考量,鋰離子電池常為第一選擇。然而,隨著儲能容量加大,電源管理加益複雜,過熱導致失火爆炸的機率增大。因此,鋰離子電池較適合用於小型的電池堆。針對容量大的儲電裝置則多考慮水力抽蓄或液流電池。前者受限於地形環境,而後者則因技術未成熟,尚於研發階段。
自然界中,已有一種金屬酵素同時具備上述相似功能:能源轉換及儲存。氫化酵素存在多數光合作用菌,藍綠藻,氮固定、乙酸、甲烷、硫酸根代謝菌中,以光合作用菌來說,水在photosystem II單元進行氧化反應產生氧氣,所代謝出的電子及質子被運送至RuBisCo單元用於二氧化碳還原,透過Calvin-Benson循環,將二氧化碳轉化為碳水化合物,完成太陽光能轉化為化學能的步驟。在過程中,多餘的電子會轉移至氫化酵素單元,以氫氣分子方式保存。此儲存的化學能也可以透過相同酵素再轉化為電流用於二氧化碳還原。也就是說,氫化酵素具備雙向功能,前一個產氫步驟為能源儲存,第二個氫分解步驟為能源轉換利用。氫化酵素因此可被視為同時具備有“鋰離子電池”和“燃料電池”功能的生物機組。
氫化酵素在進行氫氣/質子代謝上有極佳的效果,在溫和的條件下,其質子還原催化速率為每秒產生6000-9000個氫分子。在分解氫氣上,催化速率可達每秒28000氫分子。[1] 氫化酵素根據其活性中心結構來區分,可分為三類。針對化學能轉換儲存的議題來說,最佳的氫化酵素具有雙鐵活化中心。從Clostridium pasteurianum (CpI)以及Desulfovibrio desulfuricans (DdH)所分離出的活性中心,其雙鐵核心透過半胱胺酸中的硫醇官能基橋接一個四鐵四硫團簇(圖一)。在此雙鐵核心中,有一組雙硫基橋接在這兩個鐵中心上。此外,每一個鐵中心也鍵結一個一氧化碳(CO)和氰酸根(CN-)的配位基。在其中一個鐵中心,另有一個半橋接狀態的CO配位基及一個配位空位,其主要功能是允許基質配位於鐵中心進行催化反應。四鐵四硫團簇負責電子進出雙鐵中心,用以調控雙鐵核心的電子結構,以利氫氣的產生及代謝。
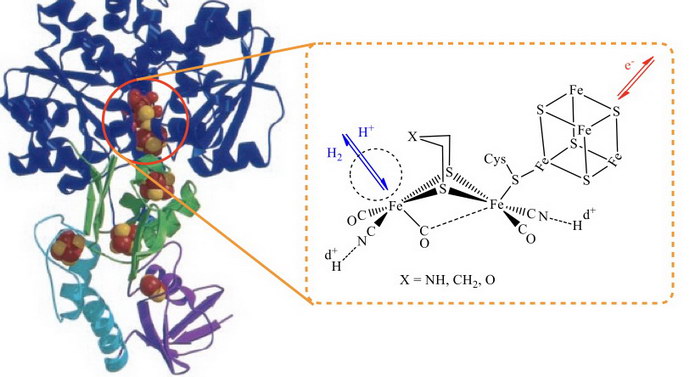
圖一 鐵鐵氫化酵素及其活化中心
在整個催化過程中,有幾個關鍵點需要釐清。第一點是,實驗室中所合成出具相似雙鐵中心的化合物,其還原電位(至少-1.3 V vs. NHE)往往高於酵素中心所量測到的催化電位(-0.5 V vs. NHE)。其次,合成化合物在還原態時,其穩定性不高,這兩項其實為合成觸媒之低催化活性之癥結所在。在我們的研究中,我們發現將具有氧化還原能力的配位基鍵結到鐵中心後,雙鐵核心的電子結構被大大影響。[2-4] 化合物的前沿分子軌域(frontier molecular orbitals)中除了鐵、硫的參與外,配位基也佔顯著的比例。這導致氧化還原反應後,電子密度並非侷限在部分金屬或配位基上,反而未定域於兩個單元上。這可紓解雙鐵核心中電子缺乏或過剩的狀態,因此穩定催化中間體。另外,透過配位基還原電位較低之輔助,可大幅降低雙鐵活化中心所需之還原電位(大於600 mV),催化電位可降到與氫化酵素之工作電位相似。這部分在降低能耗及延長觸媒使用壽命上有顯著的貢獻。
在氫氣代謝過程中,氫氣首先鍵結到鐵催化中心上,透過雙硫基上的胺基,將氫氣進行異相分解成質子和金屬氫化物。質子藉由胺基的協助,傳遞遠離雙鐵中心,鐵氫化物也會轉變為還原態鐵核及質子,後者將藉由胺基傳出鐵中心;同時,鐵核再被氧化回到起始態,等待下一次的代謝反應。在文獻中,所有實驗室中合成的相似化合物,並不具有鐵氫化合物轉變為還原態鐵核及質子的能力。由於鐵氫化合物為熱力學上最穩定的物種,要將其轉變成質子並非自發性反應。[5]在我們的研究中,匹配雙鐵核心及質子受體的pKa值為決定性的關鍵。[6] 經由適當調控雙鐵中心路易士酸鹼度,我們可以成功地引導質子轉移到質子受體上或雙鐵核心上。這種可逆反應是首次在文獻上報導的,這結果對於設計分子觸媒用於燃料電池的陽極材料有很大的幫助。
和大多數由酵素協助的催化反應一樣,調控質子和電子轉移是這類催化最重要的關鍵,例如,在Tyrosinase催化鄰苯二酚(benzene-1,2-diol)為1,2-苯醌(1,2-benzoquinone)的過程中,Tyrosinase中的酪氨酸官能基協助前者脫去兩個質子及兩個電子。[7] 一般而言,當質子化先發生在催化中心上,其還原電位就被大幅降低,或當催化中心先行接受電子,還原態的催化中心對質子的親和力就被加強;[8] 然而,欲使質子化或還原反應先發生,主要牽涉到催化核心中σ電子對強度、路易士酸鹼度,以及最低未填滿分子軌域(lowest unoccupied molecular orbital, LUMO)的能階。因此質子化和還原態反應似乎具有相互影響的因果關係,但不盡然。在某些特殊的條件下,質子和電子轉移可同時發生。[9] 在這類質子-電子偶合反應中,質子化及還原態的中間物沒有生成,反應直接略過此兩種高能階步驟,達到最終產物。在最近的研究中,我們找到一系統可進行此類反應。[10] 藉由質子-電子偶合反應,我們證明催化反應所需的能耗以及過電位大幅降低,取而代之,反應速度和催化效率顯著增加(圖二左)。也就是說,質子-電子偶合反應同時提供熱力學及動力學上的益處。
酵素在經由數億年的演化過程後,發展出高催化效率、高選擇性及低能耗的系統。透過很多的研究抽絲剝繭下,酵素的這些優點及其調控的因子,逐漸被發掘出來。解開這些秘密,只是一個開端,緊接著吾等可把這些知識運用在設計人造觸媒上(圖二右),[11] 建立一個永續的經濟。
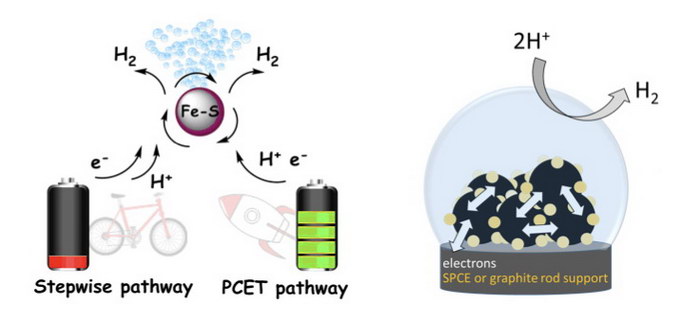
圖二(左)透過質子-電子分次反應及質子-電子偶合反應來產氫的示意圖。(右)設計含人造觸媒之電極材料用於質子/氫氣代謝反應。
參考文獻
[1] Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148.
[2] Liu, Y.-C.; Yen, T.-H.; Tseng, Y.-J.; Hu, C.-H.; Lee, G.-H.; Chiang, M.-H. Electron Delocalization from the Fullerene Attachment to the Diiron Core within the Active-Site Mimics of [FeFe]Hydrogenase. Inorg. Chem. 2012, 51, 5997–5999.
[3] Chu, K.-T.; Liu, Y.-C.; Huang, Y.-L.; Lee, G.-H.; Tseng, M.-C.; Chiang, M.-H. Redox Communication within Multinuclear Iron-Sulfur Complexes Related to Electronic Interplay in the Active Site of [FeFe]Hydrogenase. Chem. Eur. J. 2015, 21, 6852–6861.
[4] Yen, T.-H.; He, Z.-C.; Lee, G.-H.; Tseng, M.-C.; Shen, Y.-H.; Tseng, T.-W.; Liaw, W.-F.; Chiang, M.-H. Reduced Thione Ligation is Preferred over Neutral Phosphine Ligation in Diiron Biomimics Regarding Electronic Functionality: a Spectroscopic and Computational Investigation. Chem. Commun. 2017, 53, 332–335.
[5] Liu, Y.-C.; Chu, K.-T.; Jhang, R.-L.; Lee, G.-H.; Chiang, M.-H. [FeFe] Hydrogenase Active Site Modeling: a Key Intermediate Bearing a Thiolate Proton and Fe Hydride. Chem. Commun. 2013, 49, 4743–4749. (featured as a cover paper)
[6] Chu, K.-T.; Liu, Y.-C.; Huang, Y.-L.; Hsu, C.-H.; Lee, G.-H.; Chiang, M.-H. A Reversible Proton Relay Process Mediated by H-bonding Interaction in [FeFe]hydrogenase Modeling. Chem. Eur. J. 2015, 21, 10978–10982. (featured as a cover paper)
[7] Song, N.; Gagliardi, C. J.; Binstead, R. A.; Zhang, M. T.; Thorp, H.; Meyer, T. J. Role of Proton-Coupled Electron Transfer in the Redox Interconversion between Benzoquinone and Hydroquinone. J. Am. Chem. Soc. 2012, 134, 18538–18541.
[8] Liu, Y.-C.; Chu, K.-T.; Huang, Y.-L.; Hsu, C.-H.; Lee, G.-H.; Tseng, M.-C.; Chiang, M.-H. Protonation/Reduction of Carbonyl-Rich Diiron Complexes and the Direct Observation of Triprotonated Species: Insights into the Electrocatalytic Mechanism of Hydrogen Formation. ACS Catal. 2016, 6, 2559–2576.
[9] Warren, J. J.; Tronic, T. A.; Mayer, J. M. Thermochemistry of Proton-coupled Electron Transfer Reagents and Its Implications. Chem. Rev. 2010, 110, 6961–7001.
[10] Chu, K.-T.; Liu, Y.-C.; Chung, M.-W.; Poerwoprajitno, A. R.; Lee, G.-H.; Chiang, M.-H. Energy-Efficient Hydrogen Evolution by FeS Electrocatalysts: Mechanistic Investigations. Inorg. Chem. 2018, 57, 7620–7630.
[11] Chung, M.-W.; Liu, Y.-C.; Yen, T.-H.; Chiang, M.-H. Bilayer Vesicles as a Noncovalent Immobilization Platform of Electrocatalysts for Energy Conversion in Neutral Aqueous Media. ChemElectroChem 2018, 5, 20–24. (featured as a cover paper)