
蛋白質是怎麼來的呢?
蛋白質是什麼?能吃嗎?蛋白質,是一種生物巨分子。蛋白質是生物賴以維生的重要物質,其英文「protein」是 19 世紀瑞典化學家 Jöns Jacob Berzelius 所發明的 1。 Berzelius 根據荷蘭科學家 Gerardus Johannes Mulder 分析蛋白質的組成是由碳、氫、氧、氮、硫與磷的共同組合,並以希臘字「πρώτειος(proteios)」為字源,意即主要(primary),定義為「protein」。
而蛋白質的中文命名,顧名思義是跟雞蛋中的「蛋白」有關,一顆雞蛋的蛋白是由 9 成的水分與 1 成的蛋白質組成,可見蛋白質在生理上的重要性。蛋白質與核酸都是生物體內重要的成分。分子生物學的中心理論提出遺傳密碼會從 DNA 轉錄到 RNA,再從 RNA 轉譯到蛋白質。人類基因組定序於千禧年初完成後,估計人的染色體含有 2 萬多種基因,意即人類的蛋白質約有 2 萬餘種。這 2 萬餘種蛋白質是不同的,但具有相同的組合成分:胺基酸。絕大部分的蛋白質都是由 20 種必須胺基酸依照不同的序列與組成比例搭建。
蛋白質的結構與摺疊
蛋白質的序列長度不一,短的如「泛素」(76 顆胺基酸),長則如「脂肪酸合成酶」(2511 顆胺基酸),但它們都由共同的胺基酸單元組合而成。胺基酸的序列組成會影響蛋白質三維摺疊外型,也跟功能有關。這些由「胺基酸序列」引導摺疊的蛋白質在細胞內、血管中、皮膚等處都有著特殊的功能。負責蛋白質摺疊的地方也很重要!蛋白質的胺基酸序列在一種稱為「核糖體(Ribosome)」的巨型生物合成酶中即時從 RNA 轉譯,並同時進行摺疊(圖一)。
維持一個細胞的生存需要很多不同的酵素製造、催化以及分解胞內的種種化學反應。因此很多蛋白質都是生物酶,負責執行不同的功能,諸如新生胺基酸合成、核酸單元的合成、蛋白質降解、細胞內代謝小分子的合成與水解等,都跟蛋白質酶有關。蛋白質的三維造型有千千百百種不同。除擔任生物酶的功能外,有部分的蛋白質是生物骨架,負責搭架起細胞的 3 度立體空間。有些蛋白質是負責與上下游的蛋白質溝通,擔任訊號傳遞分子。有些蛋白質坐落在細胞膜內,擔任傳送化學或生物分子的向外傳送或向內運補的角色。
蛋白質被製造後,並非永久存在細胞內。細胞有一個回收廠稱為蛋白酶體(Proteasome),專門處理「用不到」、「摺疊錯誤」的蛋白質,把蛋白質拆解成一個個的胺基酸,回收胺基酸再利用。那蛋白酶體怎麼知道哪些蛋白質是要被「處理」的呢?細胞內蛋白質會被一個稱為「泛素(ubiquitin)」的小蛋白質所標記,如同急難救助現場多檢傷分類,目前已知有多種泛素的記號,不同的泛素記號影響著細胞內的生理功能。比方說有負責訊號傳遞的 K63 鍵結,DNA 修復的 K11 或 K29 鍵結。而 K48 鍵結版本的泛素鏈則跟蛋白酶體高度相關。待回收或摺疊錯誤的蛋白質經過 K48 鍵結的泛素鏈標記後,會被帶往蛋白酶體進行一系列的降解程序(圖一)。
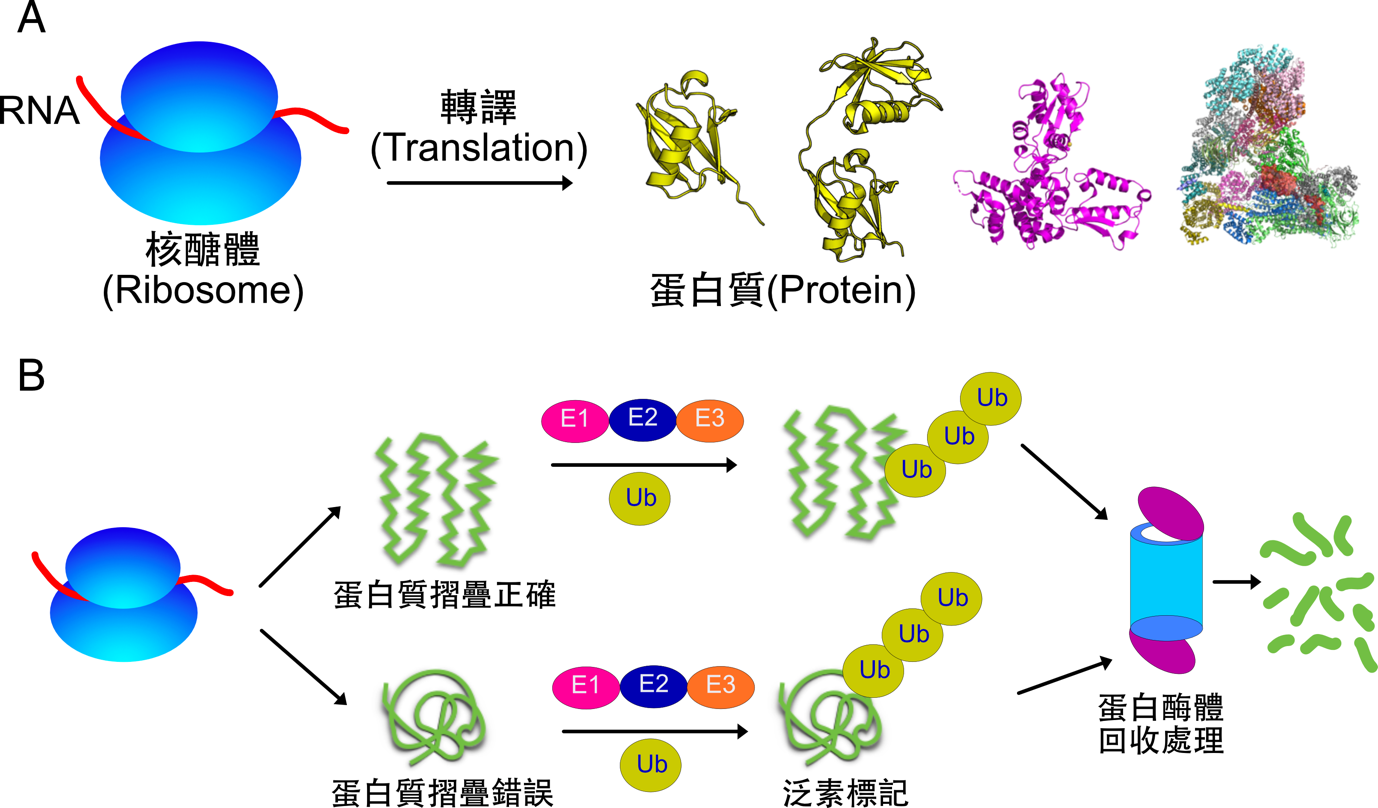
▲圖一:(A)蛋白質是從核醣體合成,並經過一系列的摺疊過程而組成一個蛋白質的立體構型。蛋白質有大有小,有些也會聚合形成一個複合體。(B)摺疊正確或是錯誤的蛋白質最終都會經過泛素的標記,送往蛋白酶體分解。
蛋白質設計在抗疾病的應用——以泛素為例
生物系統設計了泛素當作一種特殊標記符號。「標記」泛素的是很複雜的三層次連鎖酵素反應,首先由 E1 酵素啟動反應,E2 酵素轉移泛素,E3 酵素把泛素標記到目標蛋白質上。人類基因組共有超過 700 種 E1、E2、E3 酵素。當標記的功效結束後呢?生物系統也設計有「移除」泛素標記(deubiquitylation)功能的去泛素化酶。把附加的標籤(泛素)拿掉,恢復蛋白質本身的功能。這種動態的胞內泛素平衡深深的影響細胞的正常功能。
多年來科學家們嘗試使用泛素序列與構型當作骨架,製造出變種版本的泛素 X,用以競爭胞內泛素相連的連鎖酵素反應,達到改變細胞的命運的作法 2。以去泛素化酶為例,泛素鏈被去泛素化酶抓住並切除,釋放一個一個泛素回到細胞內。當修改版的泛素 X 跟去泛素化酶的親和力優化上千倍後,去泛素化酶只會被泛素 X 緊密著黏住,無法正常執行本身的功能,細胞內大部分的泛素標記的蛋白質被接續地送到蛋白酶體進行降解,可能加速癌症細胞的凋零。
這種泛素的定向演化(Directed Evolution)往往需要繁複的實驗篩選,相當耗時。試想若針對泛素的 10 個位點(胺基酸)進行定向演化,需要從 2010 種組合中篩選到適合 X 變種,需要進行大規模的實驗,成本與人力是泛素之定向演化的應用限制。近年來的結構生物學技術推展,讓科學家們可以使用先進的物理儀器,如核磁共振儀、同步輻射加速器以及冷凍電子顯微鏡量測計算出蛋白質的三維造型,再佐以生化學家對蛋白質的功能解析,了解一個蛋白質在摺疊完成後到底做了什麼事,執行了何種功能。
結構生物學家提出利用觀測到的「去泛素化酶 – 泛素」分子間的互動網絡,循理性的修改泛素,以最少的修改達到最好的泛素 X(圖二)。以此概念,我們選定了中東嚴重呼吸症冠狀病毒(MERS)中的去泛素酶 PLpro 作為技術開發與概念驗證平台 3。過往使用噬菌體篩選法找到的泛素 X 需突變 10-20 個固定位點,經由反覆篩選找出俱高親和力的泛素變種,此泛素 X 可有效抑制 MERS 病毒的複製活性。不同於傳統的篩選法,我們首先鑑定 MERS PLpro 與泛素之間的分子互動網絡,選定關鍵的互動位點,進行泛素序列修改與使用電腦計算模擬互動網絡的變化。最終只需修改 5 個位點,就可以把泛素轉換為泛素X(文章內稱為 UbV5),此 UbV5 的親和力是泛素的 2 萬 7,500 倍,比起早先用噬菌體篩選的版本優化更多。當 UbV5 與 MERS 的 PLpro 反應後,阻止 PLpro 切除細胞內的抗病毒抵禦機制,PLpro 的活性趨近於零,病毒複製也會停止。此 UbV5 有潛力作為抗 MERS 病毒的先驅藥物。同樣的設計方法也可應用到新冠病毒與多種病毒系統(圖二A)。
無中生有的泛素 X
蛋白質的三維結構是根據胺基酸序列摺疊出來的,表示有機會從胺基酸序列的組成預測未知蛋白質的三維結構。多年來科學家努力在預測與驗證中探索蛋白質三維結構,然而這個預測的複雜度是個天文數字。以泛素舉例,開頭的甲硫胺酸(Methionine)維持不變,後續的 75 顆位點皆有 20 種變化(因為有 20 種胺基酸),總共有 2075 種排列組合。當隨機變換多個位點的胺基酸後,蛋白質是否維持一樣的三維結構在過去難以準確預測。近年來的人工智慧與深度學習軟體的優化,DeepMind 公司提供的 AlphaFold 軟體大幅度的提高預測的準確度,並把人類以及其他物種的蛋白質結構都預測一番,目前累積 48 個物種的蛋白質資料庫共 2 億個結構免費開放 4。結合西雅圖華盛頓大學的 Baker 實驗室發表的 ProteinMPNN 蛋白質序列優化軟體 5,此時蛋白質的設計與驗證可全部使用電腦模擬的方式預先篩選,當計算資源不是限制的因素時,篩選的組合可比噬菌體篩選增加數百萬倍。
依此概念,我們提出一簡易的設計流程:應用 ProteinMPNN 修改泛素與 Rsp5 E3 酶之間的互動位點,用 AlphaFold 驗證 Rsp5 與泛素 X 的結合,再挑選最合理的 20 種泛素 X 序列進行實驗驗證,找出高效的泛素 X 6(圖二B)。以維持泛素三維構型為前提,ProteinMPNN 大幅度的修改泛素的序列組成,不同於設計 PLpro 專屬的泛素 X 策略(精準突變),修改過半的序列位點是稀鬆平常的數據。我們檢驗泛素 X 與 Rsp5 的互動親和力變化,與泛素 X 是否可改變 Rsp5 E3 酶的催化能力。在這 20 個實驗的版本中,有 5 個親和力提高、催化 Rsp5 活性也增加。其中編號 R4 的泛素 X 可提升親和力約 25 倍,強化 Rsp5 E3 酶的活性 6 倍以上。R4 的蛋白質晶體結構也與泛素毫無差異,這是很令人激奮的成果。20 個實證中有 5 個泛素 X 對 Rsp5 有影響力,25% 的成功率看似不高,實則效率奇高。無需人力分析、無需建構篩選平台、電腦模擬運算速率日漸加速等優勢,約略 5-10 天可完成初步篩選。
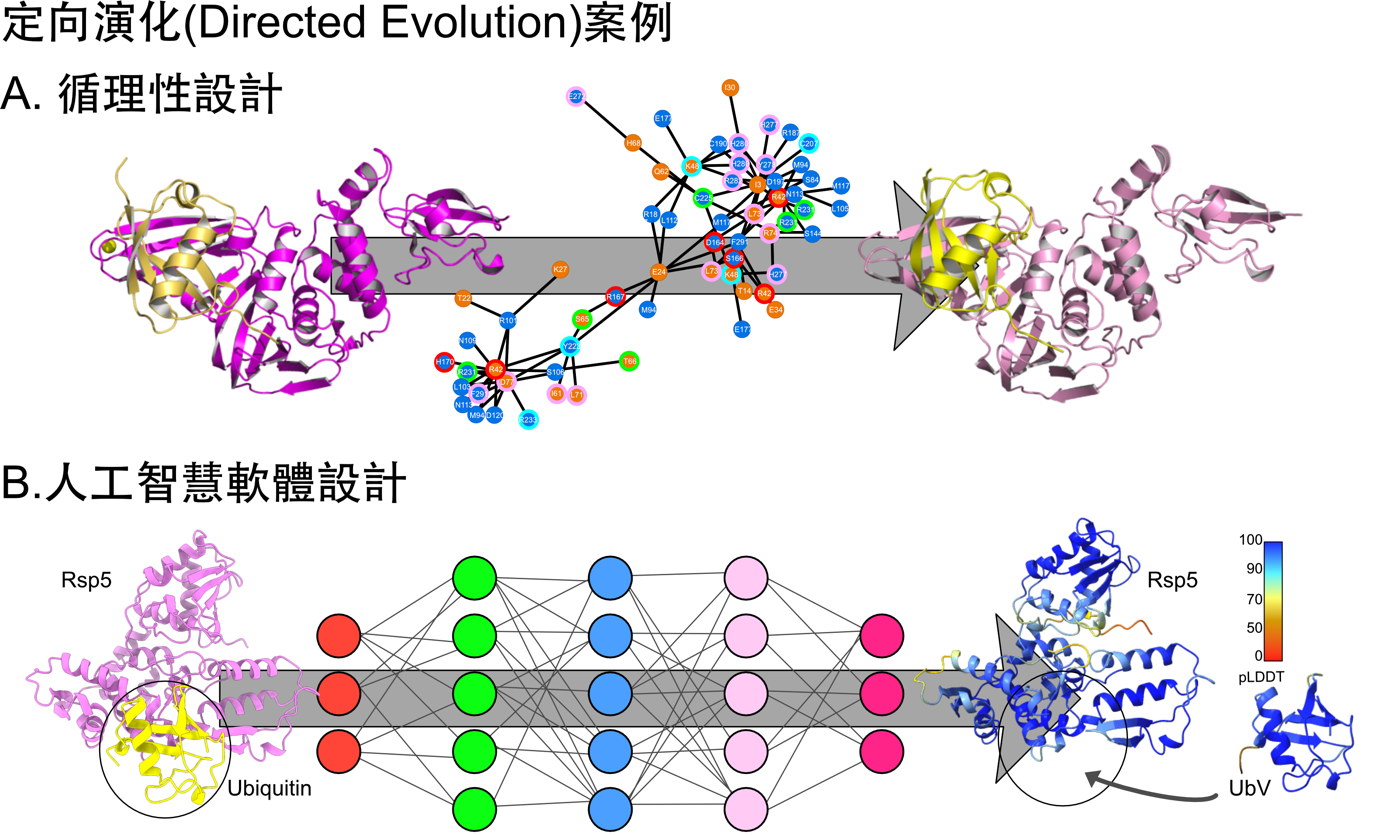
▲圖二:兩種蛋白質設計案例介紹。(A)採用循理性設計法,鑑定兩種蛋白質之間的互動網絡,優化胺基酸種類,產生新的序列。
(B)使用人工智慧軟體自動分析與生成新的蛋白質序列。
這種新穎的蛋白質設計法,將逐步取代的隨機突變定向演化法,序列修改過的蛋白質,在生技、醫藥領域的應用更是潛力無窮。
- Berzelius 提出「催化 catalysis」,「聚合物 polymer」,「異構物 isomer」等重要化學術語,他也被尊為瑞典化學之父。
- Ernst, A.; et al., A strategy for modulation of enzymes in the ubiquitin system. Science 2013, 339 (6119), 590-5. https://www.science.org/doi/10.1126/science.1230161
- Hung TI, Hsieh Y-J, Lu W-L, Wu K-P, Chang CA,“What Strengthens Protein-Protein Interactions: Analysis and Applications of Residue Correlation Networks.”BioRxiv preprint. https://doi.org/10.1101/2023.03.15.532709
- https://alphafold.ebi.ac.uk
- Dauparas, J.; et al., Robust deep learning-based protein sequence design using ProteinMPNN. Science 2022, 378 (6615), 49-56. https://www.science.org/doi/10.1126/science.add2187 Source code: https://github.com/dauparas/ProteinMPNN
- Kao H-W; et. al. Robust design of effective allosteric activators for Rsp5 E3 ligase using the machine-learning tool ProteinMPNN ACS synthetic biology 2023, online pub first, https://doi.org/10.1021/acssynbio.3c00042